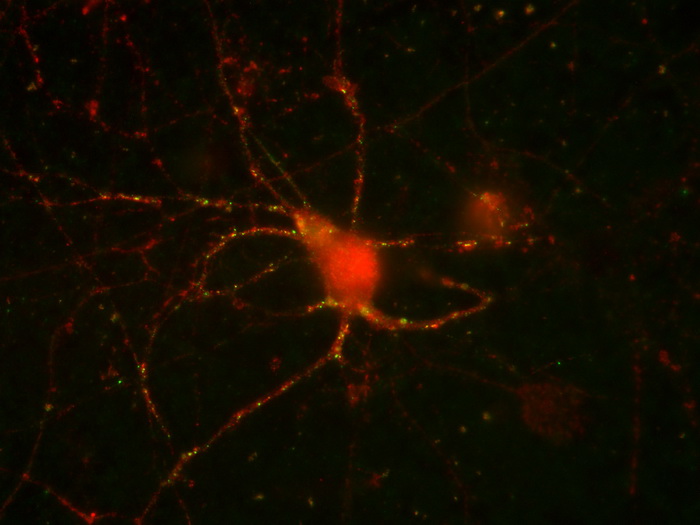
Токсичная форма белка, из-за которого начинается болезнь Лу Герига, заставляет нейроны включить компенсаторные механизмы, которые работают до тех пор, пока белка остаётся не очень много.
Нейродегенеративные заболевания, такие, как болезнь Альцгеймера, болезнь Паркинсона, боковой амиотрофический склероз (или БАС, или болезнь Лу Герига), связаны с накоплением в нервных клетках токсичных белковых отложений. Обычно эти нейродегенеративные белки нужны для чего-то полезного, но иногда, как говорится, что-то начинает идти не так. Например, белок бокового амиотрофического склероза, называемый FUS, участвует в разных клеточных процессах, управляя активностью генов. Чтобы управлять активностью генов, нужно сидеть в клеточном ядре. FUS, как и любой другой белок, синтезируется в цитоплазме, но у него в молекуле есть последовательность аминокислот, обеспечивающая ему ядерную прописку: благодаря этой метке FUS может пройти в ядро.
Но в гене FUS может случиться мутация, из-за которой транспортные белки выводят его из ядра в цитоплазму. Молекулы FUS с нарушенной ядерной меткой склонны слипаться друг с другом, образуя те самые токсичные нейродегенеративные белковые отложения. Они формируются в нейронах, связанных с мышцами; постепенно развивается паралич конечностей, потом дело доходи и до других мышц, включая дыхательные.
Сотрудники Института физиологически активных веществ РАН совместно с коллегами из Института биологии гена РАН изучали БАС в виде мышиной модели: в геном мышей вставляли ген человеческого FUS, у которого в структуре не было ядерной «прописки» – то есть белок оставался в цитоплазме, где и начинал слипаться. Но у некоторых мышей этот мутантный белок синтезировался очень активно, а у других – не очень активно. У мышей с активно синтезирующимся FUS уже в возрасте четырёх месяцев в нейронах появлялись крупные белковые включения, быстро прогрессировали двигательные нарушения, развивался паралич конечностей, и вскоре они умирали. Такая картина хорошо отражает основные симптомы БАС у пациентов. А вот у животных с низким уровнем синтеза в цитоплазме нейронов белковых агрегатов не появлялось. У таких мышей не было никаких двигательных нарушений, и жили они столько же, сколько и обычные мыши без мутантного человеческого FUS.
Но ведь медленный прирост патогенного белка – это всё равно прирост, и он всё равно должен был дать о себе знать. Видимо, в нейронах в ответ на появление аномального FUS включаются некие компенсаторные механизмы, которые помогают свести на нет вред от небольших количеств FUS. В статье в Neurochemical Research говорится, что при медленном приросте аномального FUS в нейронах мышей активировались гены, участвующие в создании межклеточных контактов и формировании внеклеточного матрикса, а также гены, помогающие формироваться двигательным нейронам по мере развития нервной системы, и гены, связанные с суточными ритмами. Очевидно, компенсаторные механизмы здесь включаются на разных уровнях, и расшифровать их будет довольно непросто; но с другой стоны, если мы научимся вовремя включать их, это позволит смягчать симптомы бокового амиотрофического склероза и продлевать жизнь больным.
Читать подробнее: Наука и жизнь